새로운 International League Against Epilepsy 뇌전증 증후군 분류: 다양한 연령대 발병 뇌전증 증후군의 분류와 정의
New International League Against Epilepsy Epilepsy Syndrome Classification: Classification and Definition of Epilepsy Syndromes with Onset at a Variable Age
Article information
Trans Abstract
Although epilepsy syndromes have been recognized for over 50 years, no formally accepted International League Against Epilepsy (ILAE) classification of epilepsy syndromes has existed until recently. The ILAE Task Force on Nosology and Definitions Taskforce (2017–2021) defined an epilepsy syndrome as “a characteristic cluster of clinical and electroencephalographic features, often supported by specific etiological findings (structural, genetic, metabolic, immune, and infectious).” Syndromes can be divided into generalized, focal, combined generalized and focal epilepsy syndromes. Some syndromes can be associated with developmental and/or epileptic encephalopathy or with progressive neurological deterioration. Two etiology-specific epilepsy syndromes have seizure onset at a variable age. The diagnosis of an epilepsy syndrome is clinically important because it frequently carries prognostic and treatment implications. The goal of this review is to provide updated diagnostic criteria for the epilepsy syndromes that have a variable age of onset, based on the ILAE special report published in 2022.
소개
뇌전증 증후군(epilepsy syndrome)이 국제뇌전증퇴치연맹(International League Against Epilepsy, ILAE) 분류 체계에서 공식적으로 등장한 것은 1985년부터였으나, 그보다 훨씬 이전부터 소아소발작뇌전증(childhood absence epilepsy, CAE), West 증후군, Lennox-Gastaut 증후군 등 여러 뇌전증 증후군들이 알려져 있었다. 하지만 가장 최근 ILAE 분류 및 용어 관련 개정이 발표된 2017년까지도 뇌전증 증후군의 공식적인 정의, 진단 기준 및 분류 체계가 정립되어 있지 않았다. 이에 ILAE에서는 2017–2021년에 새로운 질병 분류와 정의에 대한 대책위원회(Nosology and Definitions Task Force)를 구성하여 이를 정립하기 위해 노력하였다.1 그 결과로 2022년 6월 Epilepsia (volume 63)에 특별 기고로 방법론(methodology)1 이외 4개의 뇌전증 증후군 관련 성명서를 소개하였는데(신생아 및 영유아기 발병2, 소아기 발병3, 다양한 연령대 발병4, 특발 전신뇌전증[idiopathic generalized epilepsies, IGEs]5), 본 종설에서는 이 중에서 다양한 연령대 발병 뇌전증 증후군에 관한 내용을 중점적으로 요약하여 소개하려고 한다.
1. 정의1
뇌전증 증후군은 “종종 특유의 원인(구조, 유전, 대사, 면역, 감염)으로 설명되는, 임상 양상과 뇌파(electroencephalography, EEG)에서 특징적인 소견을 공유하는 집합체(cluster)”라고 정의한다. 뇌전증 증후군을 진단하면 예후 예측과 치료에도 적용할 수 있기 때문에, 가능한 경우 이를 진단하는 것이 중요한 의미를 가진다.
2. 다양한 연령대 발병 뇌전증 증후군의 분류4 (Fig. 1)
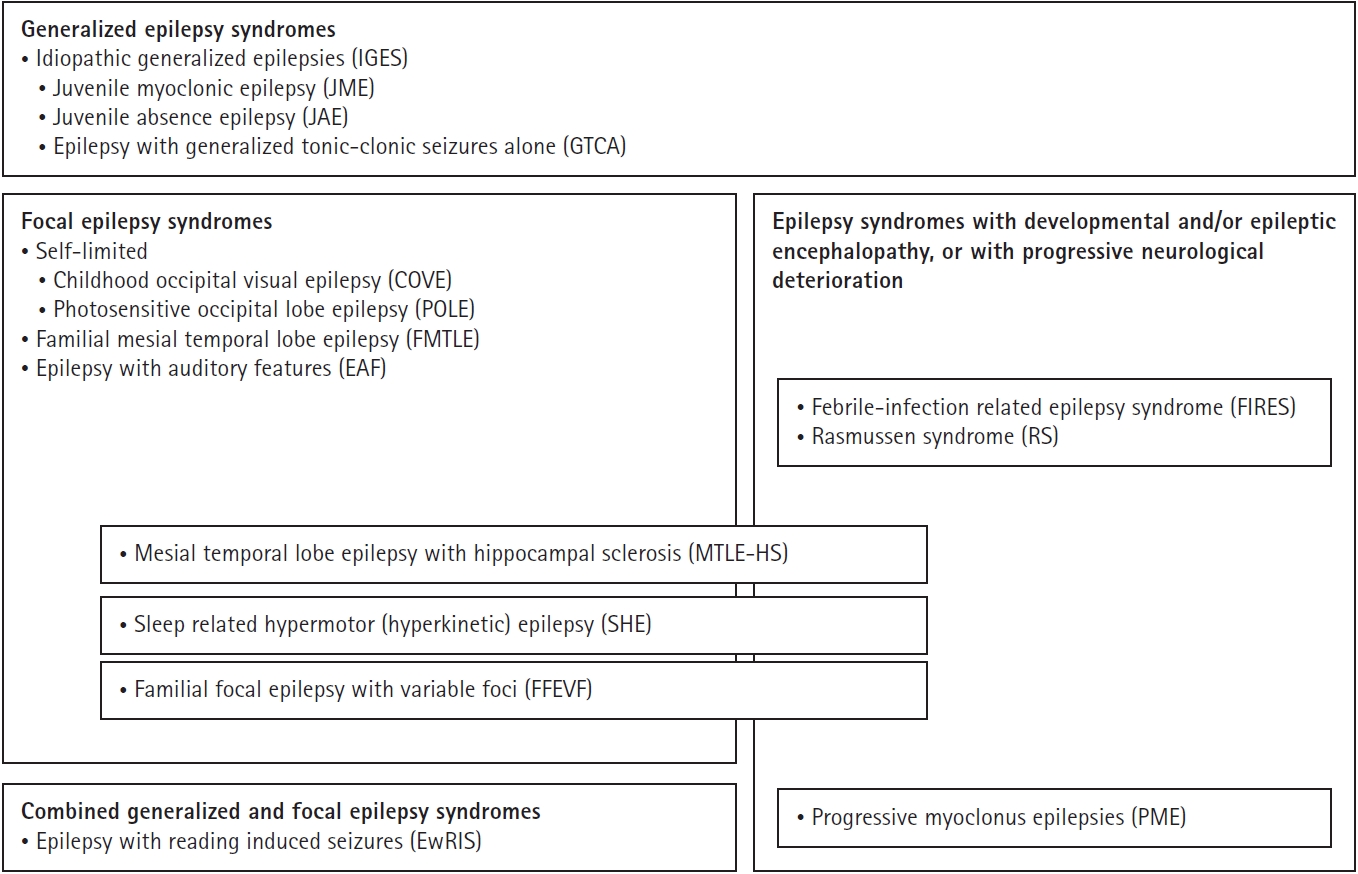
The epilepsy syndromes that begin at a variable age grouped by epilepsy type and whether they are associated with developmental and/or epileptic encephalopathy (D and/or EE) or progressive neurological deterioration. Adapted from Riney et al.4 (Epilepsia 2022;63:1443-1474) according to the Creative Commons License.
뇌전증 증후군은 흔히 나이 의존적 발생 양상을 보이는데, 본 종설에서 다루는 다양한 연령대라 함은 18세 이하와 19세 이상에서 모두 발생할 수 있는, 즉 소아청소년기와 성인기에서 모두 발생할 수 있는 증후군을 말한다.
• 전신뇌전증 증후군/다유전자(polygenic) 원인: 3개의 IGEs—청소년소발작뇌전증(juvenile absence epilepsy, JAE), 청소년근간대뇌전증(juvenile myoclonic epilepsy, JME), 전신강직간대발작만 보이는 뇌전증(epilepsy with generalized tonic-clonic seizures alone, GTCA)
• 자연 치유되는(self-limited) 국소뇌전증 증후군/복합적 유전 추정(presumed complex inheritance): 소아후두시각뇌전증(childhood occipital visual epilepsy, COVE), 광민감후두엽뇌전증(photosensitive occipital lobe epilepsy, POLE)
• 국소뇌전증 증후군/유전, 구조, 또는 유전-구조적 원인: 수면관련 과운동뇌전증(sleep-related hypermotor or hyperkinetic epilepsy, SHE), 가족 내측두엽뇌전증(familial mesial temporal lobe epilepsy, FMTLE), 다양한 초점을 동반한 가족 국소뇌전증(familial focal epilepsy with variable foci, FFEVF), 청각 특징을 동반한 뇌전증(epilepsy with auditory features, EAF)
• 전신-국소 복합(combined generalized and focal) 뇌전증 증후군/다유전자 원인: 읽기 유발 발작을 동반한 뇌전증(epilepsy with reading-induced seizures, EwRIS)
• 발달 그리고/또는 뇌전증 뇌병증(developmental and/or epileptic encephalopathy, D and/or EE)을 동반한 뇌전증 증후군과 진행하는 신경학적 악화(progressive neurological deterioration)를 동반한 뇌전증 증후군: 진행근간대뇌전증(progressive myoclonus epilepsies, PMEs), 열성 감염과 관련된 뇌전증 증후군(febrile infection-related epilepsy syndromes, FIRES)
• 원인-특이적(etiology-specific) 뇌전증 증후군: 해마경화증을 동반한 내측두엽뇌전증(mesial temporal lobe epilepsy with hippocampal sclerosis, MTLE-HS), Rasmussen 증후군(Rasmussen syndrome, RS)
상기 분류는 서로 배타적이지 않고 유연하게 적용된다. 예를 들어, KCNT1 유전자 변이로 인한 SHE의 경우 뇌전증으로 인한 신경인지 저하를 보이면 DE를 동반한 뇌전증 증후군으로 간주할 수 있다. RS 또는 MTLE-HS는 성공적인 뇌전증 수술 후 신경인지 저하가 호전되면 EE를 동반한 뇌전증 증후군으로 분류할 수 있다. 또한 PME의 경우에는 진행하는 신경학적 악화를 보이기 이전 질환 경과의 초반에는 JME와 구분이 어려울 수 있다.
3. 진단 기준 표의 구성1
각각 뇌전증 증후군의 진단 기준은 표로 정리되어 있는데, 주요 구성 항목은 아래와 같다.
• 필수(mandatory): 증후군을 진단하기 위해 꼭 있어야 하는 기준으로, 없다면 진단할 수 없다.
• 제외(exclusionary): 증후군을 진단하기 위해 꼭 없어야 하는 기준으로, 있다면 진단할 수 없다.
• 주의(alert): 증후군을 가지고 있는 대부분의 환자에서 없지만 드물게 보일 수 있어, 이 항목이 있다고 해서 진단을 배제할 수 없지만 진단을 재고하고 추가적인 검사를 고려할 필요가 있다. 주의 항목이 더 많이 존재할수록, 진단의 확실성은 점점 떨어지게 된다.
이외 일부 특정 뇌전증 증후군에서 아래 2개의 추가적인 개념을 정의하였다.
• 진행 중인 증후군(syndrome-in-evolution): 이 용어는 진행하는 뇌전증 경과의 초기에 사용되어야 하며, 발병 초기에는 필수 진단 기준을 만족하지 않지만 진행에 시간이 걸리는 경우 적용된다. 뇌영상에서 이상 소견이 보이기 이전의 RS 등 일부 증후군이 그 예가 될 수 있고, 모든 증후군에서 해당되지는 않는다.
• 검사 확진 없는 증후군(syndrome without laboratory confirmation): 이 용어는 의료 자원이 한정적인 지역에서만 적용되며, EEG나 자기공명영상(magnetic resonance imaging, MRI) 등 진단에 꼭 필요한 검사를 시행할 수 없는 경우 사용된다. 이러한 검사가 시행될 수 없다면 일부 증후군은 진단이 불가능할 수도 있다.
특발 전신뇌전증(idiopathic generalized epilepsies)5
IGEs는 유전 전신뇌전증(genetic generalized epilepsies)의 하위 범주로, CAE, JAE, JME, GTCA의 4가지 증후군만을 지칭한다(Fig. 2). IGEs는 전체 뇌전증의 15%–20%를 차지할 만큼 역학적 중요성을 가지므로 이번 ILAE 특별 기고에 별도로 정리되어 있고, 본 학술지의 이전 호에도 관련 종설이 수록되어 있다.6 이 중 다양한 연령대 발병 뇌전증 증후군에 포함되는 3개 증후군(JAE, JME, GTCA)의 진단 기준만 소개하면 아래와 같다.




국소 뇌전증 증후군
1. 수면관련 과운동뇌전증4
SHE는 수면 시 무리지어 발생하는 운동발작을 특징으로 한다. 발작은 시작과 끝이 급작스럽고, 주로 짧게 지속되며(2분 미만), 의식은 보존되고 상동적인 과운동성(hyperkinetic) 또는 비대칭적 근긴장 이상/강직(dystonic/tonic) 양상을 보인다. 이전 용어로는 ‘입면-야간 발작성 근긴장이상-뇌전증(hypnogenic-nocturnal paroxysmal dystonia-epilepsy)’, ‘야간 전두엽뇌전증(nocturnal frontal lobe epilepsy, NFLE)’, ‘상염색체우성(autosomal dominant) NFLE’로 지칭되는 증후군들을 통칭하며, 유전과 구조적 원인을 포함한다. 최근 문헌에서는 sleep-related “hypermotor” epilepsy라는 용어가 더 많이 쓰이고 있으나, 과도한 움직임을 동반한 국소 운동발작의 경우에는 “hyperkinetic”으로 표현하는 것이 더 적절한 것으로 보여, 대책위원회에서는 두 단어 모두 사용할 수 있도록 하였다(Table 4).

2. 가족 내측두엽뇌전증4
FMTLE는 청소년기와 성인기에 주로 발병하고 복합적인 유전 양상을 보이는 비교적 흔한 국소뇌전증 증후군이다. 내측두엽에서 기인하는 의식 소실을 동반하지 않는 국소발작이 주로 발생하며, 그 중에서도 기시감(déjà vu)이 특징적이다. MRI는 정상이고, 발작은 치료에 좋은 반응을 보인다. 다만 임상 양상은 다양할 수 있고, 일부에서는 선행하는 열성 경련의 병력과 MRI에서 해마 위축이 있으며 항경련제 치료 반응이 좋지 않다는 보고들도 있다(Table 5).

3. 가족 국소뇌전증4
FFEVF는 불완전 발현(incomplete penetrance) 상염색체 우성 유전 양상을 보이는 국소뇌전증 증후군으로, 가족 구성원들마다 다른 피질 영역(전두엽 또는 측두엽이 가장 흔함)에서 기인하는 다양한 강도(variable severity)의 국소 발작을 보이나, 개별 구성원에서는 단일한 국소발작 양상을 보이는 것을 특징으로 한다. 이전 용어로는 ‘다양한 초점을 동반한 가족 부분뇌전증(familial partial epilepsy with variable foci)’, ‘다양한 초점을 동반한 상염색체 우성 부분뇌전증(autosomal dominant partial epilepsy with variable foci)’으로 알려져 있었다. 유전과 구조적 원인을 가지며, 대부분의 경우 항경련제 치료에 잘 반응한다. 일부 국소피질이형성증(focal cortical dysplasia)을 동반한 약물 난치성 뇌전증의 경우, 뇌전증 수술로 완치를 기대할 수도 있다(Table 6).

4. 청각 특징을 동반한 뇌전증4
EAF는 청소년/성인기에 선행 병력 없이 발생하는 국소뇌전증 증후군으로, 청각 증상 그리고/또는 수용실어증(receptive aphasia)를 동반한 국소 발작이 특징이다. 드물게는 국소발작에서 양측강직-간대발작으로 진행하기도 한다. 일부 환자에서는 특정 소리에 의해 발작이 유발될 수 있다. 이전에는 ‘상염색체 우성 외측두엽뇌전증(autosomal dominant lateral TLE)’, ‘상염색체 우성 청각 특징을 동반한 부분뇌전증(autosomal dominant partial epilepsy with auditory features)’으로 지칭되었다. EAF가 가족성으로 발생하는 경우(familial EAF), 발현이 감소된(reduced-penetrance) 상염색체 우성 유전 양상을 보일 수 있다(Table 7).

원인-특이적 뇌전증 증후군
이환된 환자들의 대부분에서 보이는 명확히 정의되고 비교적 균일하며 특유한 임상 표현형 (임상 양상, 발작 종류, 동반질환, 질환 경과, 그리고/또는 특정 치료에 대한 반응)과 일관된 EEG, 신경영상 그리고/또는 유전적 연관성을 뒷받침하는 원인이 있는 경우 원인-특이적 뇌전증 증후군으로 규정한다.4 여기에는 2010년에 ‘특수뇌전증 및 수술치료가 가능한 뇌전증(constellations)’으로 분류되었던 MTLE-HS, RS, 시상하부과오종과 동반된 웃음 발작(gelastic seizures with hypothalamic hamartoma), 반경련-반신마비-뇌전증(hemiconvulsion-hemiplegia-epilepsy)이 포함되며, 다양한 연령대 발병 증후군에는 이 중 MTLE-HS와 RS의 2개가 해당된다.1 현재 RS 이외 자가면역뇌전증 등은 여기에 포함되지 않았으나, 향후 추가적인 연구에 따라 원인-특이적 뇌전증 증후군의 범위는 확장될 여지가 있다.1


전신-국소 복합 뇌전증 증후군

진행하는 신경학적 악화를 동반한 뇌전증 증후군
1. 진행근간대뇌전증4
PMEs는 다양한 유전적 원인에 의해 발생하는 드문 증후군으로, (1) 근간대경련(myoclonus), (2) 진행하는 운동과 인지 기능 저하, (3) 감각과 소뇌 징후, (4) EEG에서 이상 배경 서파(abnormal background slowing), (5) 이전 발달과 인지는 정상이었던 환자에서 발병함을 특징으로 한다. 광과민성(photosensitivity)은 많은 원인의 PMEs에서 공통적으로 보인다. 대부분의 환자에서 상염색체 열성 유전 양상의 가족력을 보이나, 산발적으로 발생할 수도 있다. Unverricht-Lundborg병, Lafora병, 신경세로이드리포푸스신증(neuronal ceroid lipofuscinosis), 미토콘드리아 질환(myoclonic epilepsy with ragged-red fibers, POLG-related disorders), 시알산증(sialidosis)이 PMEs의 대부분을 차지하고, 드물게는 치아적핵창백핵루이체위축증(dentatorubral-pallidoluysian atrophy), 청소년 헌팅턴병, 활동근간대경련-신부전증후군(action myoclonus-renal failure syndrome), 청소년 신경축삭퇴행위축(neuroaxonal dystrophy), 판토텐산키나제관련 신경퇴행(pantothenate–kinase-associated neurodegeneration), 뉴로세르핀봉입체병(neuroserpin inclusion body disease), 소멸백질병(leukoencephalopathy with vanishing white matter), 조발형 알츠하이머병, GOSR2 병원성 변이(pathogenic variant), 다운증후군과 동반된 근간대뇌전증, GM2 강글리오시드증(gangliosidoses), tetrahydrobiopterin 결핍, 비영아기신경병증성(noninfantile neuronopathic) Gaucher병, C형 Niemann-Pick병, 셀리악병(celiac disease) 등이 있다(Table 11).

결론
뇌전증 증후군은 임상 양상과 EEG에서 특징적인 소견을 공유하는 집합체로서, 종종 특유의 원인(구조, 유전, 대사, 면역, 감염)으로 설명된다. 뇌전증 증후군은 크게 전신, 국소, 전신-국소 복합으로 분류하고, 일부 증후군은 D/EE 또는 진행하는 신경학적 악화를 동반하기도 한다. 몇몇 원인-특이적 증후군도 있으며, 향후 연구 결과에 따라 일부 자가면역 뇌전증 등도 추후 여기에 포함될 수 있다. 뇌전증 증후군은 흔히 나이 의존적 발생 양상을 보이는데, 본 종설에서는 다양한 연령대에서 발병하는 증후군을 요약하였다. 뇌전증 증후군을 진단하면 예후 예측과 치료에도 적용할 수 있기 때문에, 가능한 경우 이를 진단하는 것이 중요한 의미를 가진다.
Notes
Conflicts of interest
No potential conflicts of interest relevant to this article was reported.
Funding
None.
Author contributions
All work was done by EKB.